About fetal diseases
About fetal diseases
- TOP
- Fetal Medicine
- About fetal diseases
What a CRIFM prenatal diagnosis can tell us
Prenatal diagnosis can examine most of the fetus’ entire body. Among the parts that will be explained, the following congenital abnormalities can be examined.
*Not all abnormalities and diseases can be identified. The fetus may not be visible due to the position and orientation of the fetus, the position of the mother’s uterine fibroids or placenta, etc. Mothers with a high BMI may have difficulty seeing the fetus or the results may not be correct.
- Head
Acrania, Encephalocele, Cephalocele, Craniosynostosis, Holoprosencephaly, ventricular enlargement/hydrocephalus, Agenesis of the corpus callosum, Cereberal maldevelopment, Chiari II malformation, Dandy-Walker syndrome, Joubert syndrome, rhombocephalosynapsis, cerebellar hypoplasia, microcephaly, lissencephaly, megalencephaly, unilateral megalencephaly, focal cortical dysplasia, polymicrogyria, schizencephaly, foramen magnum, intracerebral hemorrhage, intrauterine brain injury, arachnoid cyst, Vein of Galen aneurysmal malformation, brain tumor, intracerebral hemorrhage, calcified lesion in the brain, choroid plexus cyst - Spine
Spina bifida, scoliosis, spinal anomalies, Body Stalk malformation - Face
Orbital narrowing, orbital separation, ocular protrusion, microphthalmia, cataract, nasal bone dysplasia, nasal position abnormality (anophthalmia, elephantiasis), nasal cavity abnormality (single nostoril), cleft lip, alveolar cleft, cleft palate, mandibular narrowing, mandibular agenesis, ear position abnormality (auricular hypoplasia), external ear shape abnormality (auricular dysplasia) - Heart
Ventricular septal defect, double outlet right ventricle, tetralogy of Fallot, hypoplastic left heart syndrome, hypoplastic right heart syndrome, transposition of great vessels, aortic stenosis (constriction), etc. - Lung, Thorax
Pleural effusion, diaphragmatic hernia, diaphragmatic eventration, congenital cystic adenomatous malformation (CCAM, CPAM) - Abdomen
Umbilical hernia, gastrointestinal obstruction, esophageal obstruction, duodenal obstruction, anal atresia, , ovarian cyst, ruptured abdominal wall, asplenia, polysplenia, situs inversus - Extremities (bone system diseases)
Limb shortening, Thanatophoric dysplasia, Achondrodysplasia, Osteogenesis imperfecta, Hypophosphatasia, Joint abnormality, Hand and toe abnormality, Amniotic band syndrome, Clubfoot - Kidney, Bladder
Huge bladder, Lower urinary tract obstruction (LUTO) due to posterior urethral valve obstruction, Prune-berry syndrome, Ureteric duct remnant (allantoic cyst), Megacystis Microcolon Intestinal Hypoperistalsis Syndrome (MMIHS), Hydronephrosis, Polycystic dysplastic kidney, Ectopic kidney, Persistent Croaca, Hypospadias, Uundescended testis - Umbilical cord
Excessive coiling cord, Coiling cord, Single Umbilical Cord Artery, Location abnormality of umbilical cord insertion, (Marginal insertion, Velamentous insertion), Vasa previa - Placenta
Placenta previa, Subchorionic hematoma, Intraplacental giant hematoma (Bruce Mole)
Various congenital diseases
There are many types of congenital diseases of the fetus, and it is said that one in several dozen babies is born with some kind of disease. Here are some of the most common fetal congenital diseases by part.
Among these, those that are difficult to find during the fetal examinations conducted at CRIFM include ear abnormalities, small holes in the heart, and finger abnormalities.
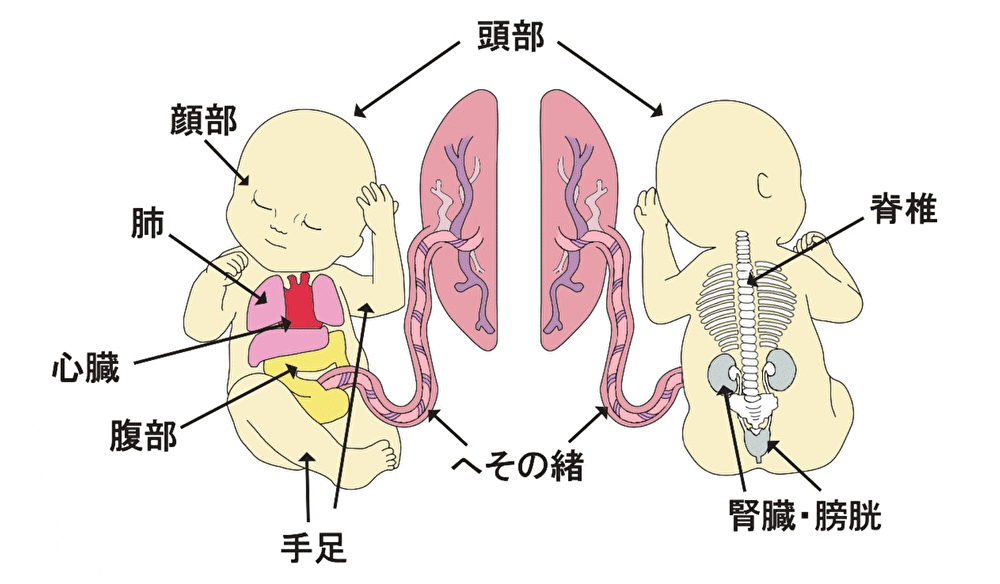
Head
1. Types of skull diseases
The fetus may have a skull that does not form properly for some reason.
Acrania
The entire skull fails to form due to failure of the neural tube to close properly.
Encephalocele
A part of the skull fails to form, causing the brain to protrude outside the skull.
Cephalocele
A so-called bump on the head, in which the brain and other tissues inside the skull do not protrude outside.
Craniosynostosis
Some diseases cause the head and face to gradually deform during pregnancy due to the fusion of the skull in the fetal stage, which is earlier than it should be. Some early skull fusion diseases are caused by genetic mutations. Complications such as facial features, syndactyly, syndactyly, and giant toes can also be seen.
2. Holoprosencephaly
The cerebrum separates into the left and right hemispheres very early in pregnancy, and the separation is clearly visible on ultrasound from about 8 weeks of pregnancy.
Holoprosencephaly
If the cerebrum does not separate properly, the disease is called Holoprosencephaly. Depending on the degree of separation, there are three types: alober, semilober and lober types. The middle part of the face is often associated with dysplasia such as cleft lip and palate, arrhinia, proboscis (elephant nose), single nostril, cyclopia or hypotelorism (orbital narrowing).
3. Enlarged ventricles and hydrocephalus
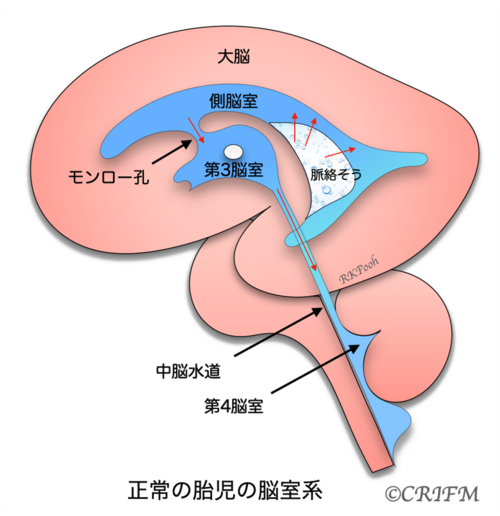
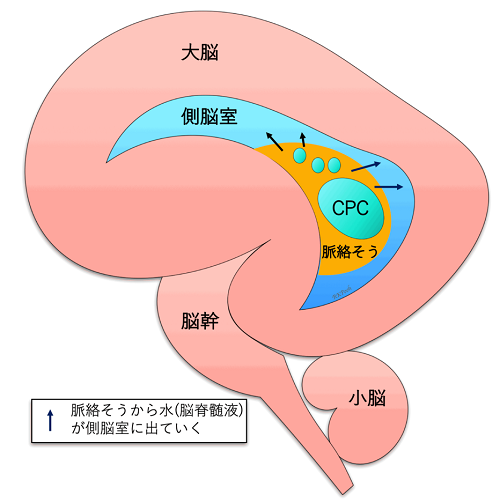
Normal anatomy
First, let’s take a look at what the ventricles of a normal baby’s brain look like. In the left and right cerebrum, water called cerebrospinal fluid is produced from the spongy part of the lateral ventricle called the choroid. The cerebrospinal fluid in the lateral ventricles flows through the foramen of Monroe into the third ventricle, then into the fourth ventricle through a small tube called the mesencephalic aqueduct, and then circulates around the cerebrum and into the arachnoid membrane around the spinal cord.
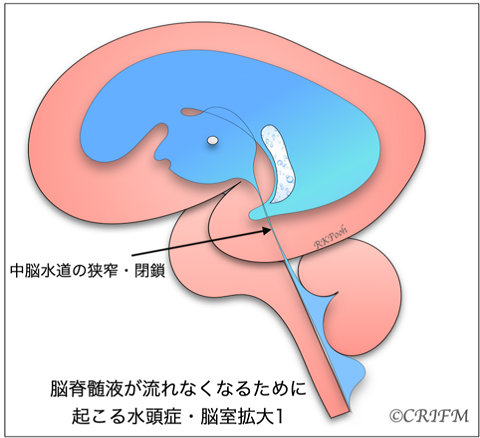
Hydrocephalus
Hydrocephalus is a condition in which cerebrospinal fluid is held back and remains in the ventricles of the brain, resulting in enlargement of the ventricles. When the “mesencephalic aqueduct” or “foramen of Monroe” in the cerebrospinal fluid pathway becomes narrow (stenosis) or clogged (closure), the ventricles above the stenosis or closure enlarge and the internal pressure gradually increases, putting pressure on the surrounding cerebrum.
When the internal pressure rises, it is called hydrocephalus, and the choroidal layer appears compressed as shown in the figure. Also, if there is a spina bifida or spinal meningioma in the back, the Chiari malformation described below may occur, narrowing the fourth ventricle and complicating hydrocephalus.
The cause of hydrocephalus can be related to various chromosomes and genes.
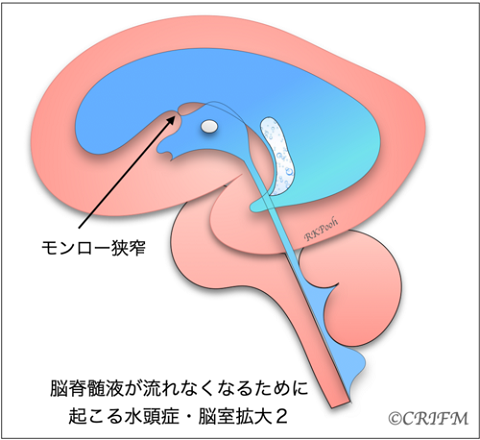
Enlarged ventricles
In a similar situation, when cerebrospinal fluid is flowing but the ventricles are large, it is not called hydrocephalus. The ventricles may be large due to the slow development of the cerebrum itself, that is, the cerebrum is thin compared to the number of weeks. In the case of the cerebral corpus callosum defect described below, the morphology of the ventricles themselves may change, causing the back of the ventricles to become larger, resulting in ventricular enlargement.
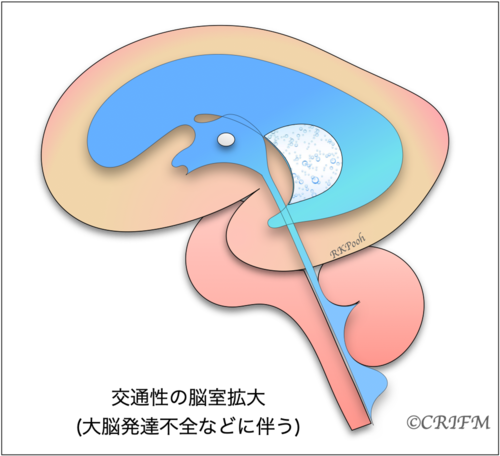
Hydrocephalus and enlarged ventricles are terms that describe a situation, not a disease. Whether it is hydrocephalus or enlarged ventricles, the cause must be clearly identified. The name of the disease depends on the cause.
In CRIFM, we narrow down the cause based on the shape of the enlarged ventricles. It is also said that there are more than 100 different genetic mutations related to ventricular enlargement and hydrocephalus. Once the causative genes of ventricular enlargement and hydrocephalus are identified, it will be possible to predict the prognosis and the degree of disability after birth.
In the case of hydrocephalus or enlarged lateral ventricles, it is important to confirm the details of the brain, not the ventricles, and to clarify the cause. Also, there may be characteristic findings on the face and fingers. In a detailed Fetal brain dock, a full body check is done to confirm the findings. Genetic testing may also be required.
In the case of hydrocephalus, treatment may include surgery to remove excess water by inserting a reservoir or shunt after birth, or surgery to allow the water in the ventricles to flow naturally, depending on the degree of hydrocephalus.
At CRIFM, babies with enlarged ventricles and hydrocephalus undergo a detailed Fetal brain dock, systemic confirmation, and genetic testing, and are referred and coordinated to the most appropriate facility depending on the cause.
4. Abnormalities of the corpus callosum
The nerve fibers that connect the right brain to the left brain are called the corpus callosum.
Agenesis of the corpus callosum
If the fetal brain has agenesis of the corpus callosum, it is necessary to conduct a detailed examination to check for other abnormalities in other parts of the brain or extra-brain complications. In the case of a corpus callosum agenesis, the shape of the ventricles changes and ventricles enlarge. If there is no other abnormality other than a defect in the corpus callosum and no genetic mutation is found, most babies will grow up healthy and without any symptoms. In cases with agenesis of the corpus callosum, it is also common to have interhemispheric cysts (water balloon) between the right and left hemispheres of the brain. Also, lipomas can form where the corpus callosum is located, but these are benign and are not an indication for surgery. Partial loss of the corpus callosum or a thin corpus callosum may be related to various diseases.
5. Cerebellar and posterior cranial fossa abnormalities
Chiari type II malformation
In babies with spinal myelomeningocele, a “Chiari II malformation” occurs. This is a condition in which the cerebellum and medulla oblongata are pulled down into the spinal canal, thus stopping the flow of spinal fluid and causing hydrocephalus. Although it is called Chiari Type II malformation, it is not actually a malformation, nor is it a disease of the cerebellum itself.
Dandy-Walker malformation
Dandy-Walker syndrome is a rare condition that causes partial or complete loss of the cerebellar vermis and cysts in the median posterior cranial fossa, which can lead to complications such as hydrocephalus. There are very few babies who are referred to CRIFM with suspected Dandy-Walker syndrome. The cases with Dandy-Walker syndrome are often associated with chromosomal abnormalities or genetic mutations, so genetic testing is always necessary.
Joubert Syndrome
Joubert syndrome is a disease of the cilia (hair-like structures on many cells in the body that allow fluid to flow in the correct direction) throughout the body. In the brain, it is known as “Moller tooth sign” because it causes ultrasound and MRI images that look like a slit in the brainstem with the absence of cerebellar vermis.
There are currently 37 subtypes of Joubert syndrome, each involving a different genetic mutation. Many children have a very severe form of the disease. Joubert syndrome is often caused by a familial genetic mutation, and if the causative gene is known, it can be confirmed early in pregnancy with CVS.
Rhombencephalosynapsis
A disease in which the cerebellum does not divide properly into left and right parts, but forms a single mass, is called “rhombocephalosynapsis,” and is said to be a rare disease. However, it can go unnoticed at birth, and the true frequency of its occurrence is not known. Some babies have hydrocephalus, which is a serious complication of the disease, and some children grow up healthy. The related gene for this disease has not yet been discovered.
Cerebellar hypoplasia
Cerebellar hypoplasia is a common disease in babies with trisomy 18 and other conditions. The shape of the cerebellum is often small but is preserved.
6. Cerebral cortical dysplasia
Malformations of the cerebral cortex (known as MCD) can lead to a variety of disabilities, including epilepsy and developmental disorders after birth.
In cortical dysplasia, gyri and sulci on the cerebral surface are poorly formed, overly formed, or irregularly shaped, but these abnormalities are not apparent until late in pregnancy or after birth.
In CRIFM, a detailed Fetal Brain Dock is performed at 18-20 weeks of gestation to determine if there are any future abnormalities in the cerebral cortex formation. CRIFM has confirmed a large number of cortical dysplasia using fetal detailed neurosonogrphy and genetic testing, although not all of them can be identified. However, the diagnosis must be made carefully, and the patient will be asked to come in for neurological follow-up scans three times over a period of 2-3 weeks to monitor the development of the brain together.
The cerebral cortex is formed through a number of steps during the first half of pregnancy. Many genes are involved in each step, and it is thought that there are more than 100 genes involved.
- Step 1: The process of growing more and more nerve cells and losing those that are not needed.
- Step 2: Neurons migrate to the surface of the cerebrum, creating a clean, layered cortex.
- Step 3: Neuronal connections are formed and neurotransmission is enhanced.
Abnormalities in these three processes are 1), 2), and 3) below.
(1) Microcephaly, microencephaly, macrocephaly, megalencephaly, unilateral megalencephaly, focal cortical dysplasia
Microcephaly is when the size of the head circumference is smaller than -3.0 SD (standard deviation) from the norm. The fetal brain dock looks at the brain through a gap in the skull called the ossicles, but microcephalic babies have a smaller gap, so the fetal brain dock may not be done properly. In such a case, a fetal brain MRI is performed to examine the situation in the brain in detail.
Macrocephaly is when the head circumference is larger than the norm by more than +3.0 SD (standard deviation). It could be a benign case where the head is simply too big for the family, but it could also be a disease caused by a problem in the initial process called megalencephaly, where brain cells overgrow or do not properly eliminate cells that are not needed. In addition, there is a case of unilateral megalencephaly, in which only one side of the brain develops, possibly due to the proliferation of cells with genetic mutations in the brain.
Localized cortical dysplasia is a partial dysplasia in the brain, which is also reported to be caused by a combination of genetic mutations.
(2) Lissencephaly
Lissencephaly is often understood as the absence of gyli and sulci on the surface of the brain, making it slippery, but there is more to it than that. There are several types of lissencephaly, which are characterized by thickening of the cerebral cortex, little or no brain gyrus, and abnormalities in brain gyrus formation.
It is classified according to the type of genetic mutation. It was believed that lissencephaly could not be diagnosed until gyli and sulci appeared in the second half of pregnancy, but CRIFM reported that early detection of lissencephaly is possible by measuring the angle of the dramatically changing sylvian fissure and observing the gap between the cerebral hemispheres and the situation around the ventricles. It is extremely important to observe whether the brain is developing according to the number of weeks in the mid-term fetal brain dock.
(3) Polymicrogyria, Schizencephaly
Polymicrogyria is a very heterogeneous cortical malformation caused by both genetic and non-genetic causes. Genetic causes include chromosomal mutations such as 22q11 deletions, 1p36 deletions and single gene mutations.
Schizencephaly is a brain abnormality that causes fissures on one or both sides of the cerebral hemispheres. It is a congenital abnormality of brain formation with traffic between the subarachnoid space and the ventricles, and the cortex at the site of the lesion often presents with multiple cerebellar gyri. Specific genetic mutations may be the cause.
It can be difficult to distinguish from porencephaly. Porencephaly is a congenital abnormality in which cysts or cavities with ventricular traffic are seen in the cerebral hemispheres. It can be caused by inflammatory diseases (infections), cerebral circulation disorders such as infarction and hemorrhage, and has also been reported to be caused by specific genetic mutations.
7. Others
Intrauterine brain damage
Intrauterine brain damage can be unpredictable, but it can occur when there is an underlying disease such as a vascular malformation in the brain, when the other baby has brain damage after the intrauterine death of a single child in a monochorionic twin, or when there is a change in blood flow. In some cases, brain damage may be found in a baby who happens to come to a mid-term checkup. If brain damage is found, we will try to find the cause of the brain damage and take measures for delivery.
Arachnoid Cysts
Arachnoid cysts are cysts (water balloons) that develop in various places inside the skull and press on the brain. They may tend to disappear during pregnancy, or they may grow in size. After the baby is born, the pediatric neurosurgery department will consider whether to operate on it.
Vein of Galen Aneurysmal Malformation (VGAM)
Vein of Galen Aneurysmal Malformation is a mass of blood vessels in the brain caused by a congenital vascular abnormality. Although it is called “varicose veins,” it is actually an arteriovenous shunt (a condition in which arteries and veins are connected without passing through peripheral blood vessels). There is a treatment for VGAM after birth. As the brain damage may occur more frequently during fetal life due to circulation problems in the brain, it is necessary to conduct neuroimaging to check the brain in detail on a regular basis,.
Other abnormalities that can be found in a fetal brain dock include the following
Brain tumor
There are various types of brain tumors, and it is necessary to diagnose whether they are malignant or benign, and also to determine the degree of bleeding associated with the tumor.
Intracerebral hemorrhage
A genetic mutation that predisposes to bleeding may be found, and maternal platelet antibodies may also play a role. In the case of intracerebral hemorrhage, the situation can change from moment to moment. Therefore, CRIFM repeats brain follow-up scans every few days or a week when brain hemorrhage is suspected, and conducts genetic tests and blood tests. In cases of intracerebral hemorrhage, CRIFM collaborates with delivery facilities to repeat Fetal brain dock.
Intracerebral calcified lesions
The presence of intracerebral calcification may be completely benign, or it may be caused by a maternal infection (cytomegalovirus or toxoplasma) that is transmitted to the baby, resulting in multiple calcifications in the brain.
Choroid plexus cyst
Choroid plexus cysts are located in the lateral ventricles of the cerebrum, where water (cerebrospinal fluid) is produced. The choroid plexus is not the part of central nerves, but just a “sponge” that produces water.
When the water coming out of the sponge accumulates inside the sponge and becomes a “cyst” (puddle), it is called a choroid plexus cyst (CPC). It is not known exactly why they form, but they are not harmful to the brain nerves.
Choroid plexus cysts are often seen in babies with trisomy 18.
The choroid plexus cysts usually disappear by about 25 weeks, but even if they do not disappear, there is no problem at all and they do not affect the development of the brain nerves. In fetal diagnosis, a detailed fetal brain dock of the baby with choroid plexus cysts is performed, brain development is observed, the general condition is checked, and the possibility of chromosome number abnormalities is confirmed. Some babies have 18 trisomy mosaicism.
Some babies have Trisomy 18 mosaicism, which is a mixture of Trisomy 18 and normal chromosomes, so if there is any concern, an amniocentesis will be performed. For Trisomy 18, you will get the definitive result the day after the amniocentesis, so you can rest assured.
Spine
Spina Bifida
The neural tube does not close properly in early pregnancy, causing the membrane that surrounds the spinal nerves and the spinal nerves to be exposed to the outside world. When the spinal nerves are exposed to the outside, it is called a myelomeningocele. When the cerebellum and medulla oblongata are pulled down into the spinal canal (Chiari type II malformation), the flow of spinal fluid is blocked, which often leads to secondary hydrocephalus.
Scoliosis
This is a condition in which the spine curves to the left or right. It includes spinal deformities seen in childhood. It is often not noticeable during fetal life, but in cases where the vertebral bodies are asymmetrically triangular in shape (hemivertebrae), the scoliosis may be noticeable from the fetal stage.
Abnormalities of the spine
Abnormalities in vertebral body formation may cause scoliosis, rib abnormalities, etc.
Body Stalk Malformation
There is no proper Japanese translation for Body Stark deformity. A Body Stark deformity is a defect in the abdominal wall, causing the abdominal organs to develop outside of the baby’s amnion, and is associated with severe scoliosis.
The scoliosis is convex on the side of the abdominal organs, sometimes the vertebra becomes ‘J’ shape, and the hips are next to the face, where the legs may appear to grow toward the head. In most cases, the craniofacial region and the upper limbs appear completely normal. The abdominal organs appear to be attached directly to the placenta. The umbilical cord is very short and the baby is unable to move. Due to the severity of the defect, this condition is almost always fatal to the fetus.
It is often referred to as “baby is too small from the beginning” or “baby’s hips do not measure up nicely.
The cause is not known, but it is highly unlikely to be repeated in the next pregnancy.
Facial
1. Eye abnormalities
Narrow orbit (Hypotelorism)
A narrowing of the space between the eyes, such as that seen in total anterior encephalocele.
Orbital deviation (Hypertelorism)
A situation in which the eyes are separated by a wide space between them, such as in chromosomal abnormalities or premature skull fusion.
Ocular protrusion (Exophthalmos)
A condition in which the eye protrudes. It is seen in abnormalities of the facial bones and premature skull fusion.
Microphthalmia
This is a disease in which the eyeball is small, also seen in trisomy 13. The function of the eye is also often lost.
Cataracts
Although it is often thought of as a disease of adults, congenital cataracts can be found in the fetus. Chromosomal abnormalities, craniofacial malformations, musculoskeletal abnormalities, and viral infections such as rubella are found in 1 to 6 out of every 1,000 children. An ultrasound scan will reveal a cloudy lens.
2. Abnormalities of the nose
Dysplasia of the nasal bones
When the nasal bones ossify slowly in babies with Down syndrome or other chromosomal abnormalities, it is called nasal bone dysplasia. However, there are family and individual differences, and a small nasal bone does not mean pathologic.
Abnormal position of the nose
It may be associated with diseases such as holoprosencephaly. Arrhinia, Proboscis.
Nasal cavity abnormality
If the nose has only one nostril, the disease is called single nostril. In this case, the back of the nasal cavity is often blocked and nasal breathing may not be possible.
3. Cleft lip, cleft alveolar, and cleft palate
Cleft lip, cleft alveolar,, and cleft palate are the most common facial abnormalities and are considered multifactorial abnormalities.
Cleft lip alone, cleft lip and cleft alveolar,, cleft lip, alveolar, and palate are all present. Cleft palate alone is overlooked by prenatal ultrasound. These abnormalities may occur on their own or as part of a chromosomal abnormality or a generalized body syndrome, so if cleft lip is present, a detailed ultrasound and chromosomal examination is necessary.
The back part of the palate, called the soft palate, may only have a cleft palate. The cleft soft palate may be combined with a narrow mandible to form a disease called Pierre-Robin sequence.
4. Abnormal mandibular development (micrognathia)
If the mandible is small (mandibular narrowing), it may be related to chromosomal or genetic abnormalities. In addition to mandibular agenesis, in which the mandible does not form at all, there are other cases in which the mandible gradually improves during pregnancy.
When the mandible is small, the position of the ears, which are formed from the same part of the jaw, is also often low. This is often a congenital abnormality caused by chromosomal or genetic mutation. Children with only a small lower jaw and normal ear position may have a disease such as Pierre-Robin sequence.
5. Ear abnormalities
Low set ears
In CRIFM, the position of the ear is confirmed at the time of early fetal dock (11-13th weeks of gestation). However, by the time of mid-term fetal dock, the position may be completely normal in normal babies.
Auricular dysplasia
Abnormalities in the shape of the ear can also be found. In some cases, only one side of the ear has a small auricle. It is very difficult to identify both sides of the auricle by ultrasonography, and even in CRIFM fetal diagnosis, only one side of the auricle can be identified in many cases.
Heart
Ventricular septal defect, double outlet right ventricle, tetralogy of Fallot, hypoplastic left heart syndrome, hypoplastic right heart syndrome, transposition of great vessels, aortic stenosis (constriction), etc.
Fetal Diagnosis of Congenital Heart Disease
Congenital heart disease occurs in one out of every 100 births.
Some have minor abnormality that do not require surgery. However, severe congenital heart disease, which requires further treatment such as surgery after birth, occurs in four out of every 1,000 births.
The purpose of fetal diagnosis of congenital heart disease is to diagnose severe congenital heart disease in utero and to be prepared to deal with emergencies that can be expected after birth.
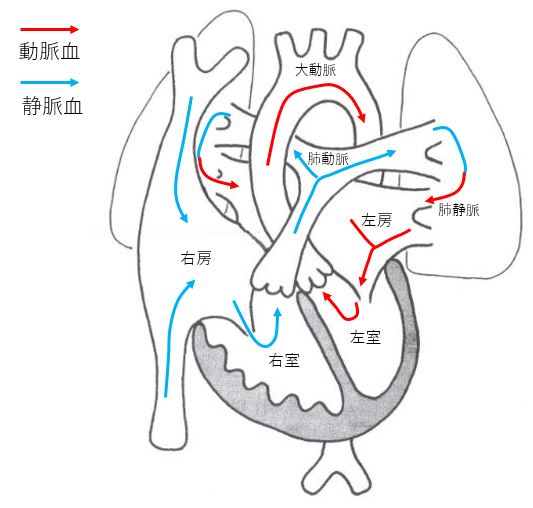
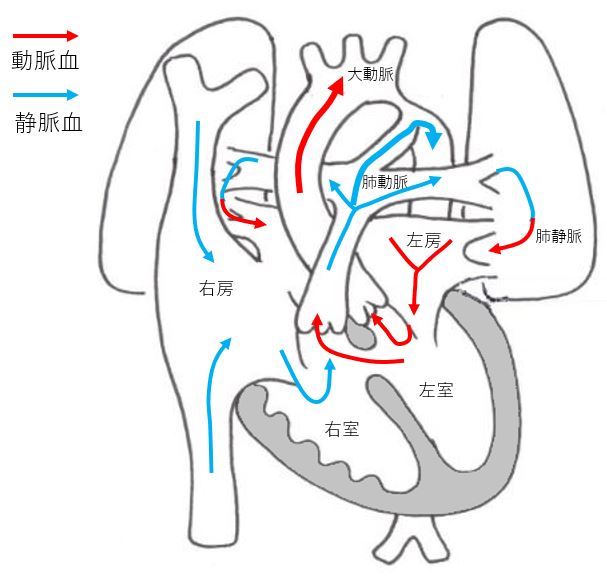
Normal heart
Ventricular septal depression (VSD)
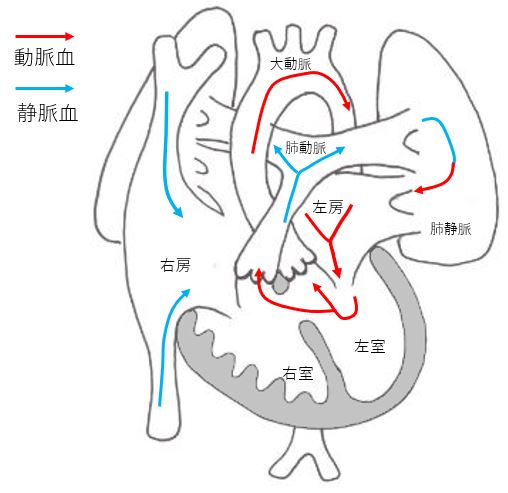
It is the most common form of congenital heart disease. It is a heart disease that leaves a hole in the wall that separates the left and right ventricles. The size of the hole determines the symptoms, but the hole may become smaller during pregnancy and after birth, and may close spontaneously. For this reason, it may be diagnosed as normal or not serious after birth.
Other heart diseases and chromosomal abnormalities may complicate the diagnosis, so tests other than those for the defective hole are necessary. Coarctation of the aorta, in particular, can cause serious complications after birth and requires specialized treatment.
If there are no abnormalities other than the ventricular septal defect, the condition does not worsen after birth. Although the frequency is high, prenatal diagnosis is sometimes surprisingly difficult.
Double outlet right ventricle (DORV)
This is a congenital heart disease in which there is a large ventricular septal defect and both great vessels (pulmonary artery and aorta) originate from the right ventricle. It is a congenital heart disease caused by an abnormality in the early stage of the ventricular septum development process. For this reason, it is sometimes difficult to distinguish it from ventricular septal defect. Because it can be complicated by other heart diseases and chromosomal abnormalities, tests other than for the defective hole are necessary.
Tetralogy of Fallot (TOF)
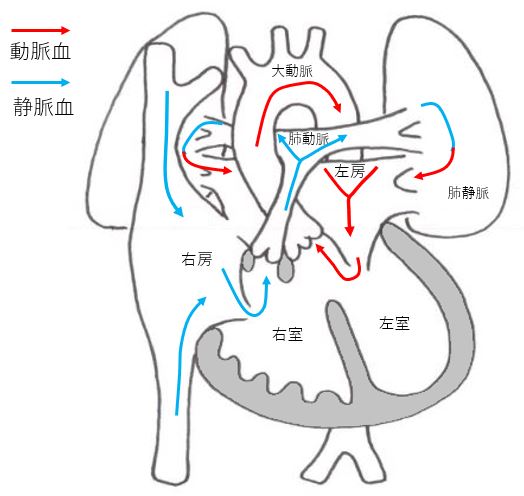
It is a typical congenital heart disease with cyanosis (pale face) as the main symptom. It is characterized by a large ventricular septum and small pulmonary arteries. The size of the pulmonary arteries affects the disease state. It is difficult to distinguish TOF from Double outlet right ventricle (DORV) because the developmental process of both diseases is the same, but there is no difference in the treatment strategy. Depending on the size of the pulmonary artery, the disease condition and treatment plan may vary, and it may be complicated by other congenital diseases or chromosomal abnormalities.
Hypoplastic left heart syndrome(HLHS)
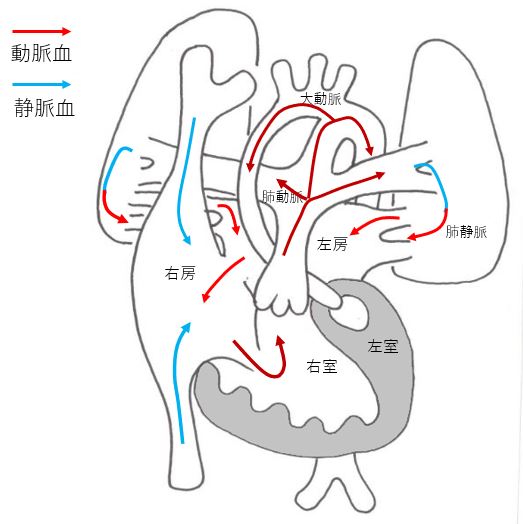
HLHS is a congenital disease in which the left ventricle, which pumps blood throughout the body, is too small to function. The blood supply to the whole body is via the ductus arteriosus. It is a serious heart disease that requires three or more surgeries, but treatment outcomes have improved over the years, with some facilities achieving a success rate of over 90%. Patients need to stay in the hospital for a long period of time and require hospital visits and medication even after the surgery.
After radical surgery, patients may be able to lead a normal daily life.
There are many cases where the patient can participate in school sports, but there are also cases with learning disabilities and needs a supportive school.
Hypoplastic right heart syndrome (HRHS)
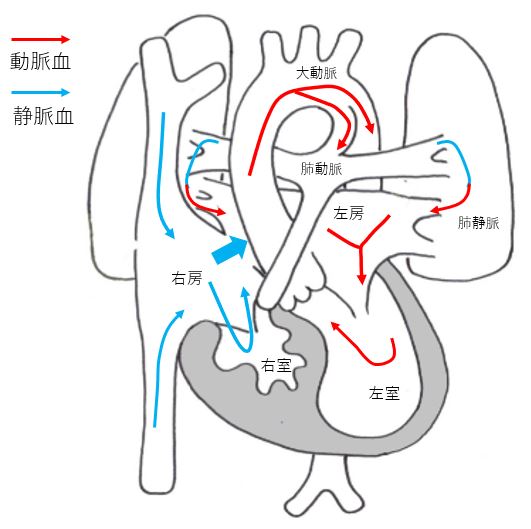
HRHS is a congenital heart disease in which the right ventricle, tricuspid valve and pulmonary artery are too narrow to supply blood to the lungs. Blood supply to the lungs is via the ductus arteriosus. In the first two weeks of life, worsening of cyanosis due to closure of the ductus arteriosus becomes a problem, requiring specialized treatment.
However, some facilities have a success rate of over 90%. After the surgery, patients still need to go to the hospital and take medication, but after the radical surgery, they may be able to lead a normal daily life. There are many cases where patients can participate in school sports.
Transposition of the Great Arteries (TGA)
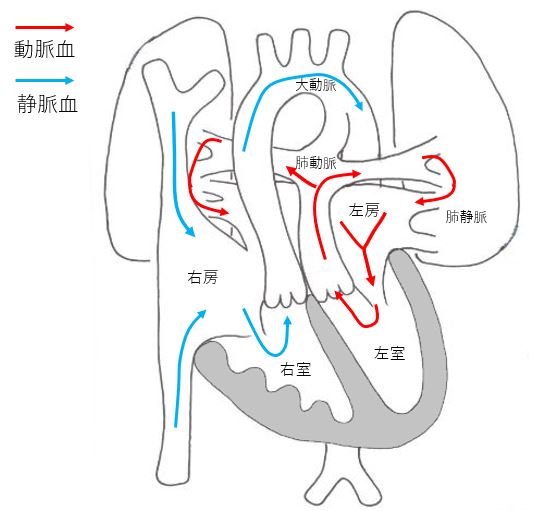
TGA is a congenital disease in which the aorta and pulmonary artery are reversed. The right ventricle ejects low-oxygen blood into the aorta, while the left ventricle ejects high-oxygen blood into the pulmonary artery. This is a severe congenital disease that causes severe cyanosis immediately after birth. It requires specialized treatment from the neonatal stage.
The treatment consists of surgery to convert the great vessels within one to two weeks after birth. The results of treatment are good, and there are many cases in which patients are able to participate in school sports after the surgery and may be able to lead a normal daily life.
Aortic stenosis (Coactation)
This is a congenital disease in which there is a narrowing of the aorta, the passage that supplies blood to the entire body. The blood is supplied to the lower part of the body through the ductus arteriosus.
There are cases of sudden onset of shock symptoms due to closure of the ductus arteriosus within two weeks of birth. Specialized treatment is necessary. However, it is not always possible to accurately diagnose the degree of narrowing. In some cases, surgery is required in the neonatal period, but in others, the stenosis thickens spontaneously. Complications of ventricular septal defect can cause severe symptoms, so careful diagnosis is necessary. Prenatal diagnosis is often very difficult.
When heart disease is suspected, abnormalities in chromosome number (aneuploidy), chromosome structural abberation and microchromosome abnormalities are highly likely. CRIFM’s CVS / Amniocentesis can confirm even microchromosomal abnormalities, so you can rest assured.
Lung and Thorax
Pleural effusion, diaphragmatic hernia, diaphragmatic eventration, congenital cystic adenomatous malformation (CCAM, CPAM)
Pleural effusion
A disease in which fluid accumulates around the lungs. It can be unilateral or bilateral. If it is only on one side, it is often a benign effusion and may disappear in 1-2 weeks. The bilateral effusion may also disappear, but if the general condition is bad, or if there is heart failure, the effusion may increase in volume.
Diaphragmatic hernia
This is a disease in which a hole in the diaphragm allows the stomach, intestinal tract and other abdominal organs to enter the rib cage, compressing the lungs and causing lung hypoplasia. The most common type of diaphragmatic hernia is a hole in the back left side of the baby’s diaphragm (Bochdalek type), but there can also be a hole on the right side or a diaphragmatic defect where most of the diaphragm is missing. In a diaphragmatic hernia, the stomach bubble and intestinal tract are often contained in the rib cage, but the most important thing is whether the liver is up in the rib cage and how small the lungs become. These are measured by various indicators in specialized facilities to make some prediction as to whether the prognosis is good or not. During delivery, it is necessary to have the baby in a perinatal center where respiratory management can be ensured from the moment the baby is born, because the smaller lungs may be collapsed if the baby makes a grunting sound. Even if the lungs are small, they will be able to function well if they are nurtured with care. The prognosis for diaphragmatic hernia after birth is very different depending on whether prenatal diagnosis is done well or not, and on whether the liver is up or not.
Diaphragmatic Eventration
Diaphragmatic eventration is a condition in which the abdominal organs (mostly gastrointestinal organs) are up to be contained in the rib cage, but in reality, the diaphragm is only loose and there is no hole in the diaphragm. Babies with diaphragmatic eventration are sometimes referred to as diaphragmatic hernias and sometimes as normal, which can be very unsettling for the patient, but the prognosis is usually good.
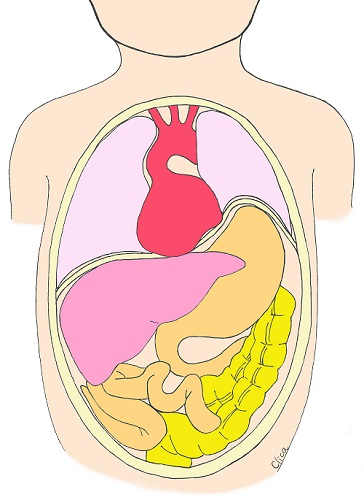
Congenital Cystic Adenomatous Malformation (CCAM) / Congenital Pulmonary Airway Malformation (CPAM)
Congenital cystic adenomatoid malformation (CCAM or CPAM) is a disease in which many cysts form in the lungs and the cyst formation causes hypoplasia of the lungs. There is a possibility of spontaneous resolution.
CCAM has been classified into three types according to the size of the cysts, but recently, a new classification (type 5) has been recommended that takes into account the histopathological site of development, and is also called congenital pulmonary airway malformation (CPAM). The size of the tumor varies, and the tumor often increases in size during pregnancy. They often become noticeable around 18 weeks of pregnancy or later and are not found in early pregnancy. If it grows significantly during pregnancy, it pushes down on the diaphragm, causing the normal lungs to be pushed down and become smaller. In severe cases, circulation may be compromised and the baby may not survive in the belly due to fetal hydrops. However, CCAM and CPAM often become smaller and smaller during pregnancy. There are many children who have been diagnosed with CCAM and CPAM but have progressively improved, or have grown even after surgery after birth, and there are many seniors at CRIFM who are now in elementary and high school.
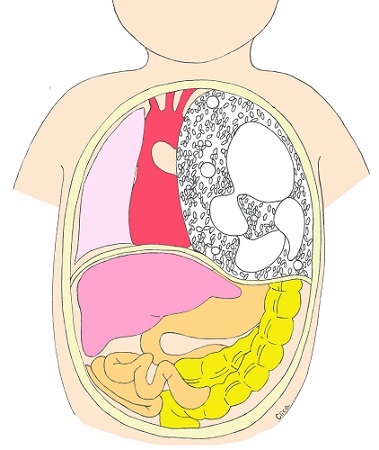
Abdomen
If we suspect that your baby has an abdominal condition, we will refer you to a specialist pediatric surgeon for prenatal counseling.
Umbilical Hernia
This is a condition in which an organ in the abdominal cavity protrudes into the umbilical cord. The protruding organ is encased in a hernia sac. It is a disease that often has complications. In the case of umbilical cord hernias, it is necessary to check for chromosomal abnormalities, and CRIFM can safely check for microchromosomal abnormalities.
Digestive tract obstruction
The fetus drinks amniotic fluid. The digestive tract includes the mouth, esophagus, stomach, duodenum, small intestine (jejunum to ileum, large intestine (colon to rectum), and anus. Gastrointestinal obstruction is a congenital disease in which there is an unconnected part (obstruction) or narrow part (stenosis) in the middle of the digestive tract. Esophageal atresia, duodenal atresia (the most common), small intestine atresia, and anal atresia are the main diseases of gastrointestinal atresia. When the gastrointestinal tract is closed or narrowed, amniotic fluid cannot flow properly, and the gastrointestinal tract above the closed (narrowed) area may become dilated, or there may be an increase in amniotic fluid, resulting in a condition called polyhydramnios.
Esophageal atresia
There are five types of esophageal atresia, from type A to type E. The most common type is type C, in which there is a tracheoesophageal fistula. The tracheoesophageal fistula allows a small amount of amniotic fluid to enter the stomach, so the stomach appears small on ultrasound and the amount of amniotic fluid increases. Complications such as chromosome number abnormalities (aneuploidy) and cardiac malformations are often seen.
Duodenal obstruction
Duodenal obstruction is the most common type of intestinal obstruction, and may be caused by chromosomal abnormalities such as Down’s syndrome (Trisomy 21), and often complications such as cardiac malformations and anal atresia are seen. In ultrasonography, the amniotic fluid in the stomach and duodenum does not flow to the rest of the gastrointestinal tract, so two dilated stomachs and duodenums stand out in the abdominal cavity, which is called the “double bubble sign”. In some cases of duodenal closure, the amniotic fluid overload (polyhydramnios) is not so obvious.
Anal atresia
Anal atresia is a congenital absence of anus that prevents the excretion of stool and is a type of recto-anal malformation. Since the fetus does not defecate in the mother’s womb, it is often found after birth when the neonate does not defecate, and is rarely found before birth. An ultrasound may show it in the early stages. Anal atresia is classified into three types; The higher the position, the more serious the malformation, and the more complicated the malformation may be. The higher the level, the more serious the malformation is considered to be, and complications may also be seen. Since it is a closure of the lowermost part of the digestive tract, the amount of amniotic fluid is usually normal.
Intraabdominal cysts
An intraabdominal cyst can have a very different course depending on where it originated. They may originate from the intestines, ovaries, or sometimes from the ureter.
Ovarian Cysts
Ovarian cysts are the most common cystic lesions in the abdominal cavity of girls. In some cases, they cause stem torsion during pregnancy, but they may shrink spontaneously.
Gastroschisis
A hole in the right side of the umbilicus causes the intestinal tract and other organs in the abdominal cavity to come out. Complications are rare, and chromosomal abnormalities and complications are rare. The prognosis is often good with postnatal surgery.
Asplenia and Polysplenia
Although people tend to think that humans are naturally left and right, there are actually parts of the human body that are asymmetrical. For example, the heart is located slightly to the left, and the stomach is also on the left. The liver is asymmetrical in shape, being larger on the right side and thinner on the left.
Asplenia and polysplenia are diseases in which the internal organs, which should be asymmetrical, are partially symmetrical. In polysplenia, the organs on the left side are bilaterally symmetrical (left isomerism), while in asplenia, the organs on the right side are bilaterally symmetrical (right isomerism ). Asplenia and polysplenia are sometimes referred to as visceral malposition. This is different from the following visceral malposition.
The structure of the heart is also asymmetrical, with the right side of the heart functioning differently from the left side of the heart. In the case of asymmetry, more than 80% of cases of asplenia and polysplenia are associated with congenital heart disease. On ultrasound examination of the fetus, the position of the stomach and the orientation of the heart should both be to the left in normal fetuses, but inconsistencies in these positions often lead to the discovery of asplenia or polysplenia. Complicated heart disease can affect the prognosis.
In addition, the spleen is involved in the immune system, which essentially protects the body, and it is said that asplenia lowers the immune function and makes the body more susceptible to infections. In cases where asplenia or polysplenia is suspected, we refer the patient to a specialist in pediatric cardiology for prenatal counseling on the condition after birth.
Situs Inversus
When the internal organs are inverted as if in a mirror, it is called Situs inversus (internal inversion). The organs of the chest and abdomen are completely inverted. In most cases, there are no complications and no need for surgery. Because the organs are completely inverted, it may not be noticed even after birth.
Extremities (Skeletal Dysplasia)
Short Limbs
More than 100 types of congenital limb shortening have been reported. Abnormalities in the formation of ribs and other parts of the body may also be seen, and the prognosis may be poor due to lung hypoplasia. The most common types of skeletal dysplasia include thanatophoric dysplasia caused by mutated growth factore gene and osteogenesis imperfecta caused by mutations of the collagen genes.
It also includes hypophosphatasia, which has recently become treatable with enzyme replacement therapy.
In addition, there are many other skeletal dysplasia such as achondroplasia, chondrodysplasia punctata, asphyxiating thoracic dysplasia, and Campomelic dysplasia.
There are various types of each disease, ranging from mild to severe, and can be detected early in pregnancy by prenatal diagnosis, or gradually become apparent after mid-pregnancy.
In this section, we will discuss Thanatophoricdysplasia, Osteogenesis Imperfecta and Hypophosphatasia.
The definitive diagnosis of these is to confirm bone findings and gene mutations. CRIFM is able to perform genetic testing for gene mutations with CVS and Amniocentesis after detailed ultrasonography, and since we are the first in Japan to provide results, many facilities refer babies suspected of limb shortening. It can also reveal mild achondroplasia and other abnormalities.
Most of the skeletal dysplasia are de novo mutations, but some are genetic disorders of parental origin. In cases of limb dysplasia, we coordinate prenatal counseling by referring the child to a geneticist or pediatric orthopedic surgeon to discuss the condition after birth.
Thanatophoric dysplasia
Tanatophrenic dysplasia is characterized by extremely short femurs and humerus from the beginning of pregnancy, and by the middle of pregnancy, shortened limbs and poorly developed ribs that give the appearance of a small, concave rib cage. It is said to be caused by mutations in a gene called FGFR3 gene, a growth factor receptor. This gene causes poor bone formation but overdevelopment of the brain, with overdevelopment confined to a part of the brain starting at about 15 weeks of pregnancy. In the fetal diagnosis of CRIFM, there is a characteristic short stature due to shortening of the limb bones and respiratory impairment due to shortening of the ribs, and many babies often die immediately after birth. In the fetal diagnosis of CRIFM, the characteristic limb morphology and abnormalities in the brain cortex are suspected from the early stage of pregnancy, and genetic testing is often used to confirm the diagnosis of Tanatophoric dysplasia.
Osteogenesis Imperfecta
Babies with osteogenesis imperfecta have fragile and weak bones that are prone to fracture and bone deformation. The sclera of the eyes may turn blue, and hearing loss may also occur. There are several types, and mutations in the type I collagen genes (COL1A1 and COL1A2 genes) are believed to be the cause. The disease can be inherited from one of the parents with the same disease, from both healthy parents with the same factor and the baby inheriting both factors, or from a mutation in which the baby has the disease while both parents have no factor. In CRIFM fetal diagnosis, osteogenesis imperfecta may be suspected early in pregnancy, and is often confirmed by genetic testing.
Hypophosphatasia
Hypophosphatasia is a disease in which a decrease in serum alkaline phosphatase (ALP) levels causes an accumulation of a substance called pyrophosphate, which impairs bone calcification, resulting in hypocalcification of bones and rickets-like changes. There are three types of the disease: mild, moderate and severe. In the severe type, the whole body bones are hardly depicted by ultrasound and the prognosis after birth is poor. In the severe form, the bones of the whole body are hardly depicted by ultrasound, and the prognosis after birth is poor. In the moderate form, the recently developed ALP enzyme replacement therapy is effective.
After birth, the condition can range from mild, with almost no symptoms, to severe, requiring constant assistance. It is suspected when the baby’s bones are crooked or there are findings of fractures, when the parents’ serum ALP levels are low, and is a definite diagnosis if there is a specific gene mutation. Although hypophosphatasia may be suspected early in pregnancy in CRIFM fetal diagnosis, it is often difficult to differentiate from osteogenesis imperfecta or other diseases in some cases. If both parents’ ALP levels are low, hypophosphatasia in the baby is suspected, but the final diagnosis is often confirmed by genetic testing. If the diagnosis of hypophosphatasia is made, we refer the baby to a geneticist for bone system diseases or a pediatric orthopedic surgeon for prenatal counseling.
Joint contractures
Wrist, elbow, and other joints may become flexed or contracted. Contracture is a condition in which a joint cannot move within its normal range for some reason. In the uterus, very low quantity of amniotic fluid (oligohydramnios) , an abnormality in the shape of the uterus and/or amniotic band syndrome can cause contractures of fetal limbs because the baby is unable to move the limbs for a long time.
In addition, babies with trisomy 18 may have contractures of the wrists from about 10 weeks of pregnancy and overlapping fingers from the middle of the pregnancy. Other chromosomal abnormalities, such as muscular dystrophy and other congenital genetic disorders, can cause the wrists, ankles, or large joints to contract and become stiff. Babies who are born with joint contractures may develop relatively normal intelligence, depending on the cause of the contracture. It is also possible to treat joint contractures after birth with various physical therapies.
Identifying the cause of joint contractures is the best way to predict the prognosis. It is necessary to elucidate the cause of the problem after detailed ultrasound and genetic testing, so that the prognosis of the baby after birth can be considered.
Hand/Foot Anomalies
Abnormalities include having too many fingers (polydactyly) or too few fingers (oligodactyly), having fingers that are attached to each other (syndactyly), and having a torn limb (split hand or split foot).
Polydactyly can be on the 5th finger side or the thumb side. Polydactyly on the 5th finger side is also seen in trisomy 13.
Polydactyly on the thumb side is also seen in Down’s syndrome, trisomy 18, and other causes. There are many different forms of polydactyly, ranging from a heart-shaped thumbnail to one with many fingers. The fetal diagnosis of CRIFM is to check the fingers and toes in the middle of pregnancy, and if there is any doubt, to take time to confirm it.
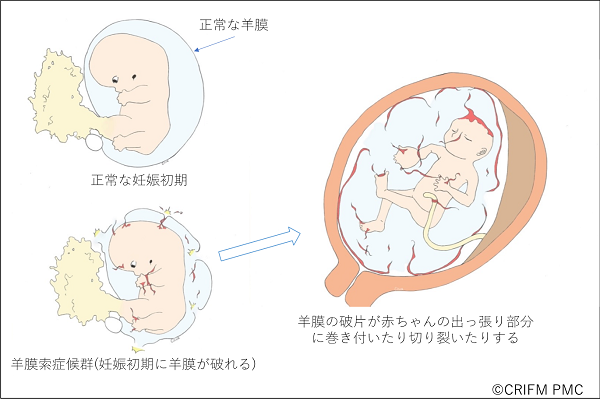
Amniotic Band Syndrome
Amniotic band syndrome is a condition in which the amniotic membrane ruptures very early in pregnancy, causing the amniotic membrane to cling to the limbs, causing parts of the body to fail to develop or to tear. This is like a car accident that occurs in the womb, and the symptoms and severity are the same as in a car accident, but are different for each individual. In some cases, only the tip of a finger is affected, but in other cases, the cranium and face are severely affected, which can be fatal.
Amniotic band syndrome is often diagnosed only by experts with considerable experience in prenatal diagnosis, except in severe cases where the cranium has been damaged and the brain has popped out. At CRIFM, we have diagnosed many babies with Amniotic band syndrome in early pregnancy.
In Amniotic band syndrome, the water balloon-like amniotic membrane that surrounds the baby ruptures very early in pregnancy, causing fragments of the amniotic membrane to damage the soft baby, thus disrupting the protruding part of the small baby. The protruding parts of the baby include the head, face, hands, feet, and umbilical cord. When the umbilical cord becomes entangled in the amniotic cord, the blood flow to the umbilical cord is subsequently compromised and the baby may die. In addition, Amniotic band syndrome is an accident that occurs outside the baby, so the organs of the abdomen and chest are usually not affected.
Diagnosis of Amniotic band syndrome, especially the tips of the hands and feet, requires time and multiple checks. This is because, unlike other congenital diseases of the fingers and toes, there is no rule of thumb for the diagnosis of Amniotic band syndrome, since it is a morphological abnormality caused by an accident.
Clubfoot
Congenital clubfoot is a condition in which a child is born with a foot that is turned inward, either unilaterally or bilaterally. It can be caused by spina bifida (spinal meningocele), chromosomal abnormalities or genetic mutations, but in many cases the cause is unknown and the condition may occur suddenly. An uncomplicated clubfoot is not noticeable in early pregnancy, but gradually develops from around 15 weeks of pregnancy, and is often found in mid-pregnancy. After birth, pediatric orthopedic surgeons can treat the condition with casts, Achilles tendon surgery, and braces, allowing the child to run and live normally. It is very important to properly determine if there are any complications.
Kidney and Bladder
Giant bladder, lower urinary tract obstruction by posterior urethral valve, Prune-berry syndrome, ureteral remnant (allantoic cyst), hydronephrosis, polycystic dysplastic kidney, ectopic kidney, migratory kidney, total excretory duct remnant
Giant bladder (Megacystis)
In early pregnancy, a large bladder is defined as a bladder longer than 7mm in diameter on ultrasound. There are many possible causes for this, as shown in the figure; chromosome number abnormalities such as trisomy 13 or 18 can also cause a large bladder in early pregnancy. Lower urinary tract obstruction (LUTO) due to posterior urethral valve obstruction, Prune-berry syndrome, and ureteral remnants (allantoic cyst), as described below, also cause a noticeable giant bladder in early pregnancy. Although uncommon.
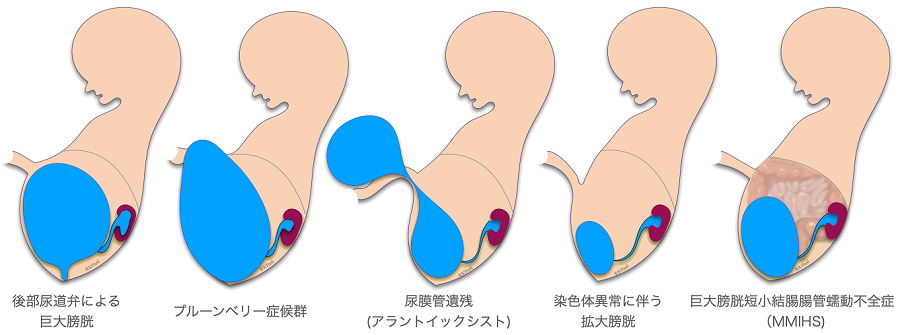
Lower urinary tract obstruction (LUTO) caused by posterior urethral valve obstruction
Urine accumulated in the bladder is excreted through the urethra and becomes amniotic fluid. The most common cause of LUTO is the obstruction of a membranous valve called the posterior urethral valve. If left untreated, both ureters and the renal pelvis will also become huge, and the kidney parenchyma will be compressed, resulting in renal dysfunction. It is an indication for fetal treatment (vesico-amniotic shunt). However, in some cases, the disease may resolve spontaneously.
Prune-belly syndrome
This is also a congenital disease that occurs mainly in boys. It shows a huge bladder, but the abdominal muscles are deficient, and there are urinary tract abnormalities and undescended testes. Prenatal ultrasound often shows a very thin abdominal surface and a protruding bladder shape as shown in the figure. There are no abdominal muscles to push urine out, but as the bladder grows to a certain extent, it naturally drains into the amniotic fluid by pressure, so the amniotic fluid volume may be maintained to some extent. Some facilities offer fetal therapy, but the complications from the missing abdominal muscles as well as the urinary tract problems can be quite severe.
Ureteral remnant (Allantoic Cyst)
In early embryonic life, the bladder is connected to the umbilical cord by the ureter to excrete early embryonic wastes to the maternal side. The ureter degenerates and becomes chordate at 4 to 6 weeks of fetal life, and the connection between the bladder and the umbilical cord disappears. However, if the ureter does not disappear, it is called ureteral remnant, and there are several types. Prenatal ultrasound shows a large bladder, a cyst that stores urine outside the body through the umbilicus, and traffic between this cyst and the bladder. In some cases, this traffic disappears during fetal life, but in most cases, after birth, when the outer cyst is removed, the urine stored in the bladder will ooze out through the navel, so surgery will be performed and steps will be taken to prevent infection. The prognosis after birth is good if there is no other associated complication.
Megacystis Microcolon Intestinal Hypoperistalsis Syndrome (MMIHS)
MMIHS is a rare disease that causes a huge bladder and short colon, and severe ileus symptoms. As the name implies, there is intestinal peristaltic dysfunction, and this condition continues after birth, making enteral feeding difficult and complete intravenous feeding common. Although it is possible to survive the fetal period with fetal treatment such as a vesico-amniotic shunt, after birth, most of the patients with this disease have a severe disease course, high mortality rate, and are considered to be eligible for small bowel transplantation or multi-organ transplantation.
Hydronephrosis
Hydronephrosis is a disease in which the renal pelvis becomes dilated and the kidney parenchyma is compressed due to ureteral stenosis or closure. It is usually unilateral, and if the other kidney is normal, it can be assumed that the kidney function and amniotic fluid volume are normal.
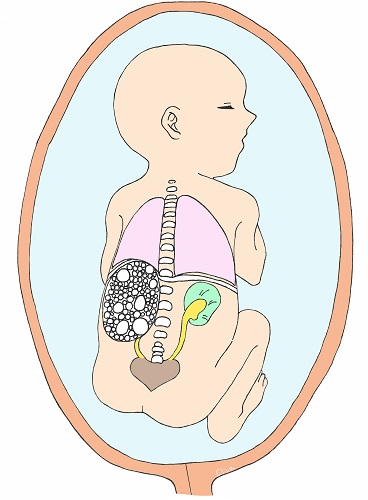
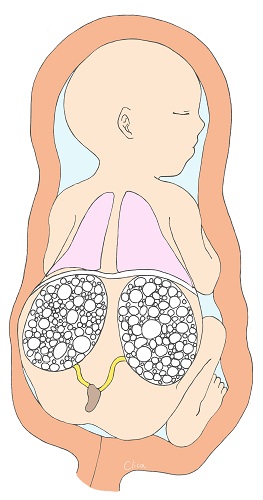
Multicystic Dysplastic Kidney (MCDK)
Multicystic Dysplastic Kidney (MCDK) is a condition in which cysts (puddles) of various sizes form within the kidney and the kidney itself becomes larger. They often shrink during fetal life. They are usually unilateral (left figure) and the prognosis is good. However, if both kidneys are affected, as shown in the figure on the right, it is bilateral renal failure, and amniotic fluid deficiency due to lack of urine production, secondary lung hypoplasia, and joint contractures in the extremities can also complicate the disease, which can be fatal.
Ectopic kidney
Ectopic kidney is an abnormality in which the kidney is not located where it should be, but is found in another part of the body (pelvis or abdominal cavity). In mid-term fetal diagnosis and late stage fetal diagnosis, both left and right kidneys are checked, which may reveal not only ectopic kidneys but also unilateral renal aplasia. Ectopic kidneys can be complicated by congenital abnormalities of the kidney, but if the other kidney is normal, the patient can lead a perfectly normal life.
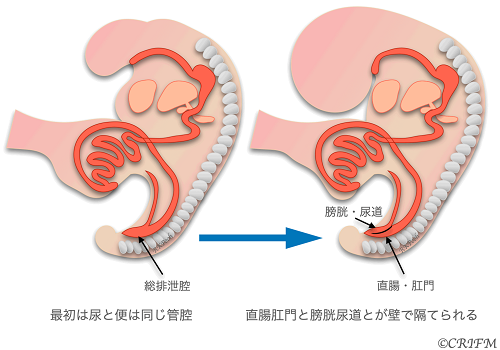
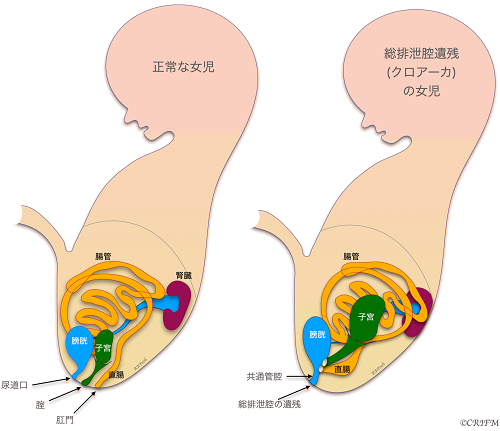
Persistent Croaca
Everyone has a cloaca at the beginning of embryonic life.
At 5 weeks of fetal life, there is no distinction between the rectum and anus and the bladder and urethra, which exist as a single cavity called the cloaca. Persistent Croaca is a disease in which the total excretory cavity is left unseparated. It occurs only in girls.
In normal cases, the urethra, vagina, and anus open into the perineum and anus, respectively, but in total voiding cavity remnant disease, the urethra, vagina, and rectum open into the total voiding cavity, and only the total voiding cavity opens into the perineum. The leftover portion of the common excretory cavity is also called the common duct. The cause of the disease is unknown, and it is believed to be caused by multiple factors, including environmental factors and genetic mutations. After birth, pediatric surgical treatment is the mainstay of treatment, but the surgery is done in stages, so as soon as the child is born, a colostomy is placed and if the valve is not mixed with urine, the child is sent home to wait for it to grow well. Then, in stages, the vagina, uterus and rectum that open into the total voiding cavity are separated, and a surgery is performed to create the rectum, anus and vagina.
Hypospadias
Hypospadias is a boys’ disease in which the exit of urine is located on the base side of the penis rather than the tip of the penis. The cause is said to be a problem with the development of the urethra or hormonal abnormalities. Complications include distended testicles and a short penis. On ultrasonography, it is suspected when the tip of the penis is slightly squared off and the penis is small. The urethral orifice can be identified by checking the direction of urination with a color Doppler at the time the fetus urinates over time, but this is not an easy task.
Uundescended testis
In boys, the testicles originate in the abdomen and descend through the inguinal canal into the scrotum. At mid-term fetal diagnosis, the testicles are not found in the scrotum in all cases, but at late stage fetal diagnosis at 29-30 weeks of gestation, both testicles are found to be properly descended into the scrotum in more than 95% of boys. Only one testicle may be present at the time of late stage fetal diagnosis, or both may be absent. If only one testicle is present, a repeat examination at about 34 weeks will almost always reveal both testicles. If both testicles are undescended, the patient may have a small penis, a hypospadias, or a chromosomal abnormality. Undescended testicles can be treated with surgery.
Umbilical cord
Excessive coiling cord
The umbilical cord is occasionally coiled tightly like the wires of a telephone handset, which can delay the baby’s growth and sometimes cause inadequate blood flow to the fetus. It may resolve spontaneously as the baby grows but may lead to intrauterine fetal demise.
Coiling cord
It is common to see the umbilical cord wrapped around the neck once or several times. It can also be wrapped around the legs or body. In most cases, this is not a problem and the cord may disappear before long. In rare cases, rotation or movement of the fetus in the uterus can cause the tangles to become stronger, resulting in intrauterine fetal death. In addition, during delivery, the umbilical cord tangles can cause poor cord circulation, resulting in low fetal heart rate and emergency cesarean section.
Single Umbilical Cord Artery
The umbilical cord usually contains three blood vessels, two arteries and one vein, but there are fetuses with only one artery, which can be associated with chromosomal abnormalities such as trisomy 18 and/or Turner syndrome. There is no need to worry about the umbilical cord after birth, as it is something that can be severed after birth.
Location abnormality of umbilical cord insertion, (Marginal insertion, Velamentous insertion)
If the umbilical cord comes out of the placenta at the edge of the placenta, or if it comes out of the membranes instead of the placenta, the baby’s growth may be slow and blood flow may be inadequate. In addition, in cases where the membranes is attached to the placenta, there is often a complication of vasa previa as described below, which needs to be properly diagnosed.
Vasa previa
When the blood vessels leading to the umbilical cord run in the wall of the uterus near the internal os, it is called a vasa previa. Vasa previa is most likely to occur when the umbilical cord is attached to the membranes. Sometimes previa can also occur when the umbilical cord is protruding from the normal position of the placenta. The blood vessels leading to the umbilical cord crawl through the membranes, so when the membranes ruptures during delivery, and these blood vessels also rupture, causing the baby’s blood to flow out rapidly, resulting in the baby’s death or severe sequelae such as cerebral palsy even if the baby manages to survive. If a baby is delivered vaginally, the fetal mortality rate can be as high as 50% or more. On the other hand, the survival rate is as high as 97% in cases of prenatally diagnosed preeclampsia.
In CRIFM, the site of attachment of the umbilical cord is checked during early fetal diagnosis at 11-13 weeks of gestation, and if the membranes are attached, it is also checked for the possibility of an anterior vascular vessel, but finally, the presence or absence of an anterior vascular vessel is confirmed during mid-term fetal diagnosis at 18-20 weeks. However, in the end, we will confirm the presence of previa at 18-20 weeks by mid-term fetal diagnosis and refer the patient to a facility that has experts in previa. So far, all patients with previa who have been referred to experts have delivered healthy babies.
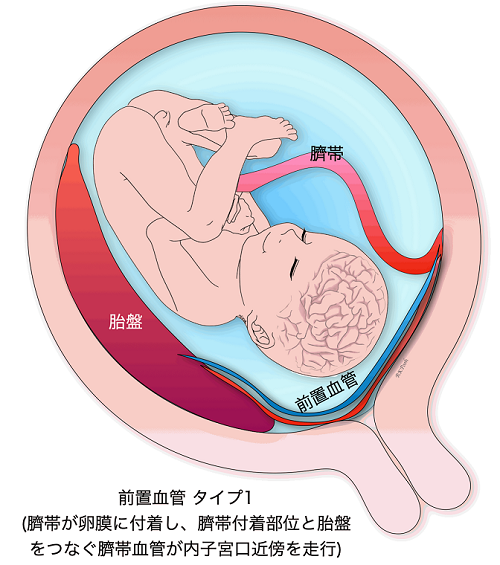
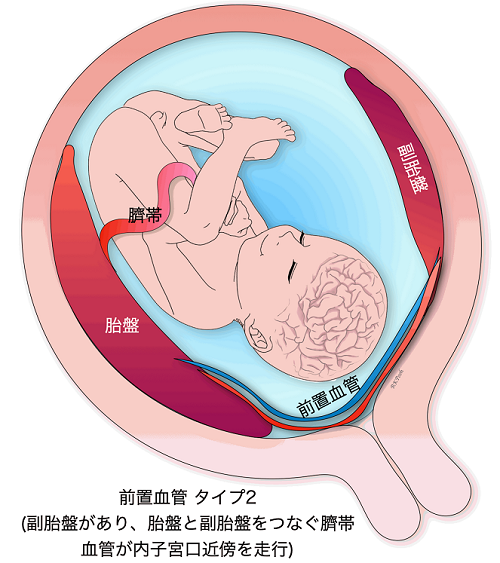
Placenta
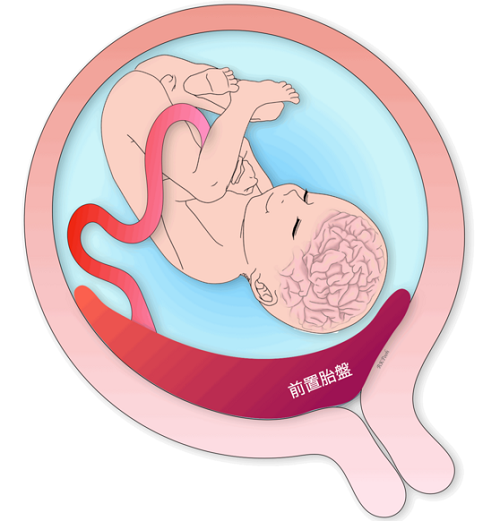
Placenta previa
Placenta previa is a condition in which the placenta attaches to a lower position than normal (closer to the vagina) and thus covers part or all of the internal os. Older pregnancies, smokers, multiparous women, twins, and previous uterine surgeries are said to be at risk for placenta previa. In addition, 5-10% of placenta previa are “placenta accreta”, which is known as placenta previa and often requires a total hysterectomy.
Subchorionic hematoma (hemorrhage)
Subchorionic hematoma is a bleeding in the uterus that is commonly seen in early pregnancy. There is no difference in the prognosis (miscarriage rate) if the bleeding is seen in early pregnancy. However, in cases of repeated intrauterine bleeding, the hematoma may become infected, leading to miscarriage in the mid-term.
Intraplacental Giant Hematoma (Breus’mole)
Intraplacental hematoma ((Breus’ mole) is a rare giant hematoma that occurs on the baby side of the placenta. This disease is caused by a giant hematoma in the placenta, which impairs the circulation of the fetal placenta and frequently leads to intrauterine fetal growth failure or intrauterine fetal death, and the prognosis of the fetus is often poor.